Review Article
The role of genetic mutations in genes LMNA, PPARG, PLIN1, AKT2, CIDEC in Köbberling–Dunnigan Syndrome

Shahin Asadi* and Mahsa Jamali
Division of Medical Genetics and Molecular Pathology Research, Tabriz, Iran
*Address for Correspondence: Shahin Asadi, Molecular Genetics, Director of the Division of Medical Genetics and Molecular Research, Tabriz, Iran, Email: [email protected]
Dates: Submitted: 08 February 2019; Approved: 21 February 2019; Published: 22 February 2019
How to cite this article: Asadi S, Jamali M. The role of genetic mutations in genes LMNA, PPARG, PLIN1, AKT2, CIDEC in Köbberling–Dunnigan Syndrome. J Genet Med Gene Ther. 2019; 2: 001-006. DOI: 10.29328/journal.jgmgt.1001004
Copyright License: © 2019 Asadi S, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Köbberling-dunnigan syndrome; LMNA; PPARG; PLIN1; AKT2; CIDEC genes; Metabolic disorder

Abstract
Köbberling-Dunnigan syndrome, also known as partial familial lipodystrophy, is a rare genetic disorder characterized by abnormal distribution of adipose tissues. Many people with Köbberling-Dunnigan syndrome develop insulin resistance, a condition in which body tissues cannot adequately respond to insulin hormone. Insulin is a hormone that helps regulate the level of your blood glucose. Köbberling-Dunnigan syndrome can be due to mutations in several different genes. However, type 2 Köbberling-Dunnigan syndrome is caused by the mutation of the LMNA gene, which is located on the long arm of chromosome 1 as 1q22.
Köbberling-Dunnigan syndrome, also known as partial familial lipodystrophy, is a rare genetic disorder characterized by abnormal distribution of adipose tissues. Adipose tissue is usually seen in many parts of the body, including under the skin and around the internal organs [1] (Figure 1).
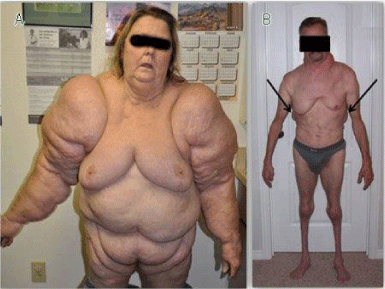
Figure 1: A woman with Köbberling-Dunnigan syndrome (left) and a male with this syndrome (right side) with abnormal distribution of adipose tissues (adipose).
Therefore, fat not stored in the organs is created around the face and neck and inside the abdomen. In people with Köbberling-Dunnigan syndrome, fat tissue is lost from the arms, legs, and pelvis, and these parts of the body appear muscular. Therefore, fat not stored in the organs is created around the face and neck and inside the abdomen. Excessive fat in these areas gives people an appearance similar to those in Cushing’s disease. This abnormal fat distribution may begin at any time from childhood to adolescence [2] (Figure 2).
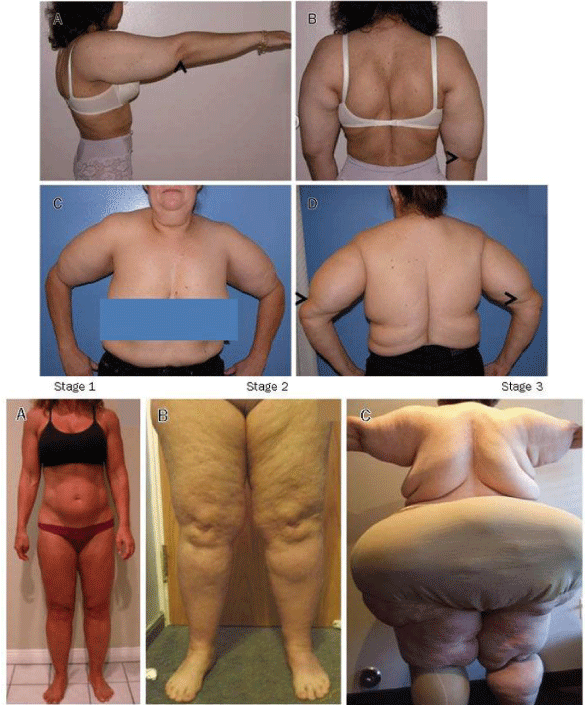
Figure 2: Another view of adipose tissue disorder in patients with Köbberling-Dunnigan syndrome.
Abnormal storage of fat in the body can lead to health problems in adulthood. Many people with Köbberling-Dunnigan syndrome develop insulin resistance, a condition in which body tissues cannot adequately respond to insulin hormone. Insulin is a hormone that helps regulate the level of your blood glucose. Insulin resistance may get worse to become more seriously called diabetes. Some people with Köbberling-Dunnigan syndrome, an abnormality of acanthosis, have a skin condition associated with high insulin levels in the bloodstream. Acanthosis nigricans causes skin scaling, wrinkling, thickening, darkening and tightening of the skin. Acanthosis nigricans is a brown to black, poorly defined, velvety hyperpigmentation of the skin. It is usually found in body folds, such as the posterior and lateral folds of the neck, the armpits, groin, navel, forehead, and other areas [3] (Figure 3).

Figure 3: Another view of a man with Köbberling-Dunnigan syndrome associated with the related disorder
Often people with Köbberling-Dunnigan syndrome also have high levels of fat called circulating triglycerides in the bloodstream (hypertriglyceridemia) which can lead to pancreatitis (inflammation of the pancreatitis) [4].
In addition, abnormal accumulation of fat in the liver (fatty liver), which can lead to liver enlargement (hepatomegaly), which can lead to impaired liver function, may also occur in the Köbberling-Dunnigan syndrome. Some women with Köbberling-Dunnigan syndrome after puberty may experience multiple cysts in the ovary, increased body hair growth (hirsutism), and infertility, possibly related to hormonal changes [5].
The researchers have described six forms of Köbberling-Dunnigan syndrome, whose genetic cause has been identified. Among them, Type 2 Köbberling-Dunnigan syndrome is the most common form, also known as Dunnigan disease. In addition, some people with Köbberling-Dunnigan syndrome may have myelopathic disorder, heart muscle impairment (cardiomyopathy), and impaired cardiac conduction [5] (Figure 4).
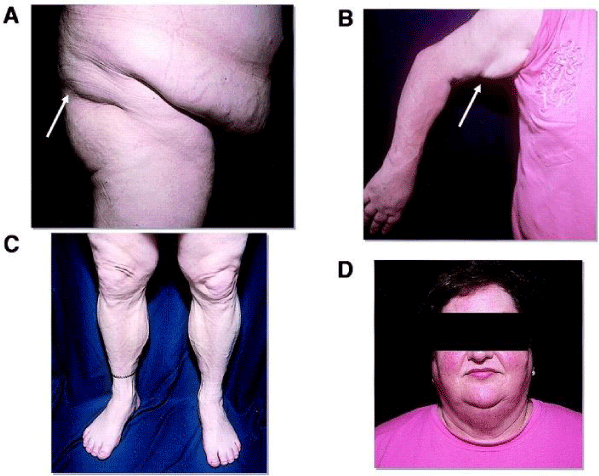
Figure 4: Other Implications of Disorders in Köbberling-Dunnigan syndrome.
Etiology of köbberling-dunnigan syndrome
Köbberling-Dunnigan syndrome can be due to mutations in several different genes. However, type 2 Köbberling-Dunnigan syndrome is caused by the mutation of the LMNA gene, which is located on the long arm of chromosome 1 as 1q22. The LMNA gene provides instructions for the synthesis of proteins that contain a variety of cell functions, including fat storage. In particular, these proteins play an important role in the development and functioning of adipocytes, which are fat storage cells in fatty tissues. The mutation in any of the genes associated with Köbberling-Dunnigan syndrome reduces or eliminates the function of the relevant proteins, which reduces the growth of the structure or function of the adipocytes, and as a result, the body can maintain and use lipids in Positions are not suitable. These disorders of the adipose tissue produce hormones and affect many organs of the body. However, it is not clear why these changes make fat stored in some parts of the body and disappear in some others. It is worth noting that no mutation has been identified in some patients with Köbberling-Dunnigan syndrome, and researchers believe that genetic or other epigenetic changes may also be involved in the development of this syndrome [6] (Figure 5).
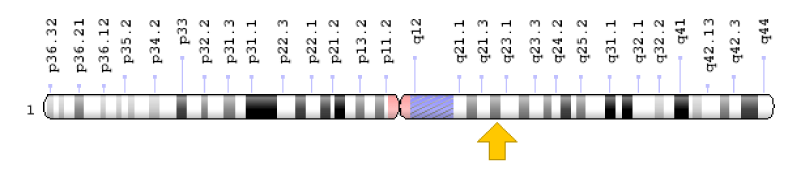
Figure 5: Schematic view of chromosome number 1, where the LMNA gene is located in the long arm of this chromosome as 1q22.
Often cases, Köbberling-Dunnigan type 2 syndromes follow the dominant autosomal inheritance pattern. Therefore, in order to produce this syndrome, a mutated version of the LMNA gene (parent or parent) is required and the chance of having a child with Köbberling-Dunnigan syndrome in the dominant autosomal state is 50% for each pregnancy [6] (Table 1).
| Table 1: Name of effective genes in various types of Köbberling-Dunnigan syndrome with chromosomal position. | ||
| Syndrome Type | Chromosome position | Gene |
| 1 | ? | ? |
| 2 | 1q22 | LMNA |
| 3 | 3p25.2 | PPARG |
| 4 | 15q26.1 | PLIN1 |
| 5 | 19q13.2 | AKT2 |
| 6 | 3p25.3 | CIDEC |
Frequency of köbberling-dunnigan syndrome
Köbberling-Dunnigan syndrome is a rare genetic disorder with an estimated 1 in 1 million live births in the world. Type 2 Köbberling-Dunnigan syndrome is the most common form of this disorder, and so far, more than 500 cases have been reported from all over the world in medical literature [7] (Figures 6-10).
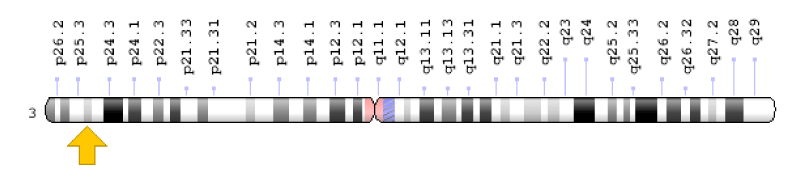
Figure 6: Schematic view of chromosome number 3, where the PPARG gene is located in the short arm of this chromosome as 3p25.2.
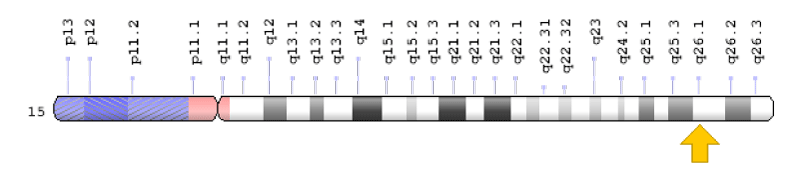
Figure 7: Schematic view of chromosome number 15, where the PLIN1 gene is located in the long arm of this chromosome as 15q26.1.

Figure 8: Schematic view of chromosome number 19, where the AKT2 gene is located in the long arm of this chromosome as 19q13.2.
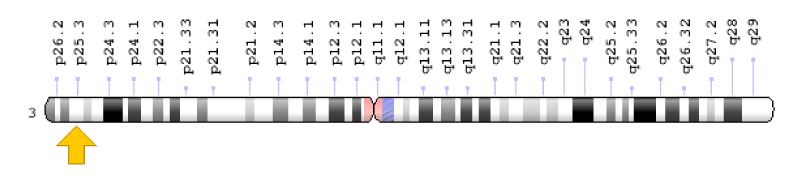
Figure 9: Schematic view of chromosome number 3, where the CIDEC gene is located in the long arm of this chromosome as 3p25.3.
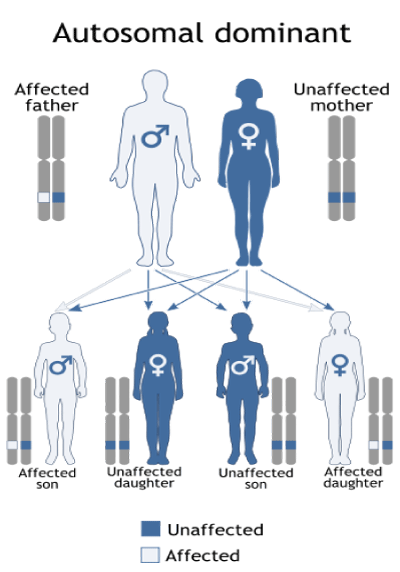
Figure 10: Schematic view of the dominant autosomal inheritance pattern that follows the Köbberling-Dunnigan syndrome.
Diagnosis of köbberling-dunnigan syndrome
Köbberling-Dunnigan syndrome is diagnosed based on the clinical and physical findings of the patients and some pathological examinations. The best way to diagnose this syndrome is to test for at least the genetic gene of LMNA to investigate the presence of possible mutations [8].
Therapeutic routes for köbberling-dunnigan syndrome
The Köbberling-Dunnigan syndrome treatment and management strategy is symptomatic and supportive. Treatment may be done by a team of experts, including gastroenterologist, liver specialist, clinical biochemist and other healthcare professionals. There is no standard treatment for this syndrome and all clinical measures are needed to reduce the suffering of the infected person. Genetic counseling is also a special place for all parents who want a healthy baby [8].
Discussion and Conclusion
Köbberling-Dunnigan syndrome, also known as partial familial lipodystrophy, is a rare genetic disorder characterized by abnormal distribution of adipose tissues. Many people with Köbberling-Dunnigan syndrome develop insulin resistance, a condition in which body tissues cannot adequately respond to insulin hormone. Insulin is a hormone that helps regulate the level of your blood glucose. Köbberling-Dunnigan syndrome can be due to mutations in several different genes. However, type 2 Köbberling-Dunnigan syndrome is caused by the mutation of the LMNA gene, which is located on the long arm of chromosome 1 as 1q22. Köbberling-Dunnigan syndrome is a rare genetic disorder with an estimated 1 in 1 million live births in the world. There is no standard treatment for this syndrome and all clinical measures are needed to reduce the suffering of the infected person.
References
- Bidault G, Vatier C, Capeau J, Vigouroux C, Béréziat V. LMNA-linked lipodystrophies: from altered fat distribution to cellular alterations. Biochem Soc Trans. 2011; 39: 1752-1757. Ref.: https://goo.gl/r1gEV3
- Garg A, Agarwal AK. Lipodystrophies: disorders of adipose tissue biology. Biochim Biophys Acta. 2009; 1791: 507-513. Ref.: https://goo.gl/uU4HBk
- Garg A. Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab. 2011; 96: 3313-3325. Ref.: https://goo.gl/s4RLDo
- Guénantin AC, Briand N, Bidault G, Afonso P, Béréziat V, et al. Nuclear envelope-related lipodystrophies. Semin Cell Dev Biol. 2014; 29: 148-157. Ref.: https://goo.gl/wqAtHS
- Rapini RP, Bolognia JL, Jorizzo JL. Dermatology. Set St Louis. Mosby. 2007; 2: 1541–1542. Ref.: https://goo.gl/FNaLrX
- James WD, Berger TG, Elston D. Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. 2006; Ref.: https://goo.gl/uVyA3c
- Herbst KL, Tannock LR, Deeb SS, Purnell JQ, Brunzell JD, et al. Köbberling type of familial partial lipodystrophy: an underrecognized syndrome. Diabetes Care. 26: 1819–1824. Ref.: https://goo.gl/cnjmZZ
- Garg A. Clinical review#: Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab. 2011; 96: 3313–3325. Ref.: https://goo.gl/Ux3tn9