Review Article
The advances and challenges of Gene Therapy for Duchenne Muscular Dystrophy

Jean-Paul Iyombe-Engembe and Jacques P Tremblay*
Centre de Recherche du CHU de Quebec-Université Laval, Quebec, Canada
*Address for Correspondence: Dr. Jacques P. Tremblay, Centre de Recherche du CHU de Quebec-Université Laval, JPT, Quebec, Canada, Email: [email protected]
Dates: Submitted: 14 June 2017; Approved: 24 July 2017; Published: 25 July 2017
How to cite this article: Iyombe-Engembe JP, Tremblay JP. The advances and challenges of Gene Therapy for Duchenne Muscular Dystrophy. J Genet Med Gene Ther. 2017; 1: 019-036. DOI: 10.29328/journal.jgmgt.1001003
Copyright License: © 2017 Iyombe-Engembe JP, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Gene therapy, Duchenne muscular dystrophy, CRISPR/Cas9, Animal model

ABSTRACT
Since the discovery of the dystrophin gene (DMD gene) thirty years ago, several therapeutic approaches have been investigated to treat Duchenne muscular dystrophy (DMD). This includes cell therapy, exon jumping, exonic knockout, and the CinDel method. In this article, we present the challenges of developping a treatment for DMD and the advances of these various approaches. We included the new CRISPR-Cas9 system, which permits not only major progress in the development of new treatments based on genome editing but also the production of new animal models.
INTRODUCTION
The last ten years have been characterized by unprecedented breakthroughs in the field of biology. This extraordinary development is the result of several achievements, one of the most significant of which was the culmination of the Human Genome Project (PGH) in 2004. This project, which lasted more than ten years and which mobilized considerable human and financial resources for sequencing the entire human genome. This gigantic project was made possible thanks to the progress of sequencing technology and bioinformatics [1].
The identification of the three billion nucleotides of the human genome obtained after sequencing the 23 chromosomes, which constitute the human genome, was only the first stage of the Human Genome Project. This is the visible part of the iceberg because the major challenge is to identify the functions of the different sequences of the human genome [2].
To address this legitimate concern, the Encyclopedia of DNA Elements Project (ENCODE) was created by the National Human Genome Research Institute (NHGRI). It aims to understand how the cell uses the information contained in the genome, the functions of all the elements coded in the human genome. The first results of the ENCODE Project show that the human genome consists of only about 2% of the coding parts and more than 98% of the non-coding parts. There would be only 20,000 genes to code about 100,000 proteins.
Genome analysis suggests that there are about 7000 genetic diseases due to modifications of the human genome. Some of these diseases are multigenic and thus affect several parts of the genome. Others are monogenic and due to a mutation of only one gene or one non-coding part of the genome.
Duchenne muscular dystrophy belongs to the latter group. It is caused by a series of mutations that affect the DMD gene coding for dystrophin located on the short arm of X chromosome [3,4]. The mutations can be punctual, translocations, duplications and especially shifts of the reading frame due to a deletion. But regardless of the type of mutation, the result is the same: absence of complete dystrophin protein in the patient’s muscle fibers [5,6]. However, since the discovery of the cause of the disease, thirty years ago, several approaches of gene therapy have been considered.
The first approach used in gene therapy against DMD was the grafting of normal myoblasts obtained from muscle biopsies of non-dystrophic persons [7]. Myoblasts were transplanted intramuscularly to the patient. After transplantation, the myoblasts can fuse with the recipient’s fibers and allow for the synthesis of normal dystrophin [8]. As will be discussed later, this approach has a number of advantages and disadvantages.
The second approach is to deliver the normal DMD gene to the muscle fibers with a viral vector. But this approach was abandoned because there were no viral vectors that could contain 2.4 Mb of the complete dystrophin gene. The other variants of this approach, such as the delivery of the dystrophin cDNA (13 kb) with the adenoviral vector or the delivery of a short cDNA (4 kb) with an adeno-associated vector to produce a mini- or micro-dystrophin in the muscle fibers have so far failed to achieve the desired clinical results [9].
The third approach called exon skipping, which uses antisense oligonucleotides is to produce a dystrophin mRNA deleted of one or several exons to restore the reading frame shift produced by the patient deletion [10,11]. After translation, it is thus possible to obtain a dystrophin protein internally truncated, which is more or less functional. This approach was aimed at transforming a Duchenne patient with a severe phenotype into a less severe phenotype Becker patient. As will be seen in the remainder of the discussion, the exon skiping did not result in any significant improvement in the patient.
The fourth approach is using recombinant endonucleases. We distinguished two types of endonucleases: those that recognize the target sequence of DNA with a polypeptide sequence (MGNs, ZFNs, TALENs) [12-14] and the one, which uses an RNA-type polynucleotide sequence to recognize the target DNA sequence (i.e., the CRISPR/Cas9 system) [15]. Both types of endonucleases can generate DNA double-stranded breaks (DSBs), which can be repaired by Non-Homologous End Joining (NHEJ) resulting in micro-insertions or micro-deletions (INDELs) which can restore the reading frame and thereby transform a Duchenne patient into a Becker patient. The DSBs may also be repaired by Homology Directed Repair (HDR), which allows replacement of a sequence or insertion of the missing sequence. It is thus possible to restore the complete dystrophin gene and transform a young Duchenne patient into a normal individual.
We will examine the advances and difficulties encountered in recent years in gene therapy for DMD as well as the challenges to be faced.
ADVANCES AND CHALLENGES OF CELL AND GENE THERAPY FOR DMD
Advances and challenges of myogenic cell transplantation
The first trials of transplantation of myogenic cells were done about forty years [7]. Whether it is allogeneic, autogenic or even xenogeneic cells, the transplantation of myogenic cells in animal models or in humans aims to achieve a triple objective.
1) To allow fusion of transplanted myogenic cells with the host muscle fibers. The fusion with the muscle fibers is at the root of the genetic complementation phenomenon in which the myogenic cells of the donor (having the wild gene) provide a normal gene to compensate for the mutated patient gene. There is a genetic mosaicism where healthy nuclei co-exist in the same muscle fibers with the mutated nuclei [16].
2) To allow the formation by the transplanted myogenic cells of new muscle fibers, which must occupy spaces where muscle tissue has been destroyed and replaced with non-functional connective or fatty tissue [17].
3) To generate new satellite cells, the cells which are the main source to regenerate the muscle tissue following damage (Figure 1) [18,19].
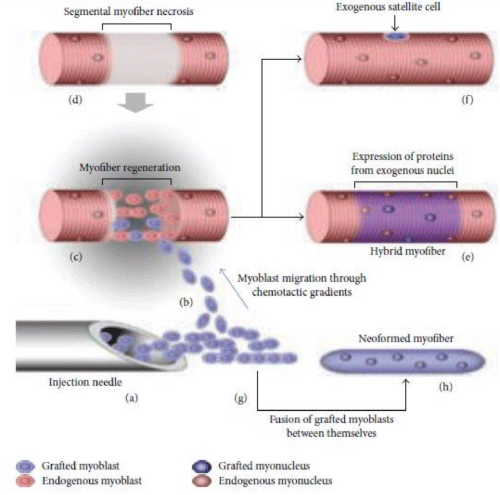
Figure 1: Diagram of transplantation of allogeneic myoblasts by intramuscular route. a) Injection of allogeneic myoblasts into the patient's muscle with a needle. b) The injected myoblasts migrate to the perimysium space due to the chemotactic gradient. c) The regeneration of part of the patient muscle fiber (blue). d) Muscle regeneration occurs in the necrotic part of the patient's muscle fiber. e) The regenerated part of the muscle fiber express proteins coded by the exogenous nuclei. f) The transplanted myoblasts also form new satellite cells. g) The grafted myoblasts can fuse together. h) This results in the formation of new muscle fibers.
It should be noted that satellite cells may not be the only myogenic cells capable of regenerating muscle tissue. Other cells such as myoendothelial cells [20], mesoangioblasts [21], pericytes [22], CD33 + [23] and recently cells with aldehyde dehydrogenase activity (ALDH) + [24], interstitial cells PW1+/Pax7- [25] and β-4-integrin+ cells [26] have also been reported to participate in muscle regeneration following their transplantation. Recent advances in cell reprogramming may also be an important source of myogenic cells obtained by the differentiation of iPSC [27].
However, to achieve this threefold objective, several obstacles have to be overcome. These include obtaining and processing the cells to be transplanted, selecting the best route of transplantation, controlling the immune response of the host following the transplantation of allogeneic or xenogeneic cells and developing methods to monitor the transplantation success. The ultimate goal of this approach is the development of a rigorous transplantation protocol in order to ensure successful transplantation on one hand and, on the other hand, to allow the recipient to benefit from it without aggravating his health. In the next section, we will examine the various points listed above.
Obtaining and processing the cells to be transplanted
Most of the myogenic cells used in transplantation are obtained from biopsies of the skeletal muscles of donors. The samples are taken by a surgeon following a protocol approved by the research ethics committee [28]. Biopsies are treated with proteolytic enzymes (collagenase and trypsin) that degrade connective tissue and release satellite cells. The latter are then cultured in vitro to ensure their proliferation as myoblasts. Two major constraints must be overcome at this stage: avoid contamination of successive cell cultures by microorganisms (bacteria, fungi, endotoxin and especially viruses) and maintain the cells at the myoblast stage avoiding their fusion as myotubes.
Selecting the best route of transplantation
There are only two routes of administration of myogenic cells that have been investigated: intramuscular and intraarterial [21,28]. Before examining the two routes of administration, it seems useful to review the pre-treatment of the host muscles before transplantation. Pre-treatment aims to improve the success rate of transplantation by promoting the migration and fusion of myogenic cells with the host muscle fibers. There are two pre-treatments that have been mainly tested: the use of phospholipases [29] and the introduction of intense muscle activity in the animal model [30]. These treatments are aimed at damaging the host muscle fibers. To remove host satellite cells in mouse models, the muscles are either irradiated or cryo-damaged [31,32].
The intramuscular route involves injecting the myogenic cells with a needle mounted on a syringe. It has the advantage of directly bringing the transplanted cells into the host muscular tissue [28,30]. This contribution can, in the best cases, allow the in situ differentiation of the myogenic cells and their fusion with the host muscle fibers located close to transplanted cells. While the approach is effective in animal models with small muscles, such as mice and rats, it is very laborious in nonhuman primates and in humans with larger muscles. Muscle size is not the only obstacle to this approach. The accessibility of certain affected muscles such as the diaphragm, a muscle involved in breathing is a limiting factor in the intramuscular route.
The systemic or intraarterial approach may, in theory, resolve most of the difficulties associated with the intramuscular route. However, it faces a major difficulty. Indeed, according to some studies, the vast majority of myogenic cells (except perhaps mesoangioblasts and CD133+ cells) cannot be extravasated after systemic administration [21,33]. This could be explained by the fact that the myogenic cells do not have a deformable cytoskeleton similar to that of the red blood cells, which can give them the capacity to circulate inside capillaries having a diameter less than their own. This would also allow myogenic cells to cross the junctions of endothelial cells at the level of the capillaries to leave the circulation and reach the muscular tissue where they must differentiate and fuse with the patient muscle fibers. This difficulty may explain the use of the intramuscular route to the detriment of the systemic pathway in the vast majority of myogenic cell transplantation trials.
Controlling the immune response of the host following the transplantation of allogeneic or xenogeneic cells
In almost all transplants, transplanted myogenic cells are allogenic (healthy mice to an mdx mouse, non-human primate to another non-human primate and healthy human to a dystrophic human) and, to a lesser extent, xenogeneic (human, dog or pig myoblasts to a mouse) [19]. In both cases, the immune system of the recipient should be either nonfunctional or permanently suppressed by immunosuppressive drugs to allow for successful transplantation. Some investigators have used cyclosporin A as an immunosuppressive drug [34]. Although immunosuppressive during cell transplants in mice, cyclosporin reduces the fusion of transplanted myoblasts with the host muscle fibers by blocking cell differentiation and inducing apoptosis at therapeutic doses.
Transplantation trials with tacrolimus (FK 506) in mice showed good fusion of myoblasts with the host muscle fibers [35]. This immunosuppressant was also used during myoblast transplantations in non-human primates and in clinical trials with excellent results [28]. To date, tacrolimus remains the only immunosuppressant used in monotherapy in transplantation of myogenic cells because of its low toxicity at therapeutic doses to the recipient and to transplanted myogenic cells. Beyond the use of immunosuppressive drugs, the development of immune tolerance in the host remains a possible alternative [36]. Several studies are being carried out in this direction to solve the problem posed by the long-term use of immunosuppressants. Immuno-deficient mice (nude, rag or SCID) have also been used for transplantation [37-40].
One solution to the problem posed by the rejection of transplanted allogeneic myogenic cells is the use of genetically corrected autogenous myogenic cells. We will address this aspect of the problem in the section on gene therapy.
Developing methods to monitor the transplantation success
The transplantation monitoring has to be done at the short and long term. At short term, the follow-up aims to evaluate the early mortality of transplanted cells. Several studies [19,28] confirmed that approximately 70-80% of transplanted myogenic cells die within 72 hours of transplantation. This high mortality, although it does not prevent transplantation as a therapeutic approach, leads to the necessity to transplant more cells to the DMD patients. The mechanisms underlying this high mortality have not yet been clearly elucidated. Myoblasts that pass from an in vitro (culture) state to an in vivo (recipient’s muscle) state are likely to experience a number of stresses that seriously affect their survival in the new environment. Some studies have identified the expression of pro-apoptic and necrotic factors (intracellular Ca2 + deposition) by transplanted cells that could explain early death by apoptosis [45]. However, these different studies did not explain the upstream factors that activate the different cell death pathways. A careful study of the microenvironment of the transplant is crucial to know the mechanisms underlying the high early mortality. The study of the microenvironment should take into account the mechanical aspects of transplantation, vascular lesions and the mechanism of in situ coagulation and the supply of nutrients to transplanted cells and the different mechanisms of cell death (apoptosis, autophagy, necrophagy...).
With regard to the mechanical aspects, the diameter of the needle lumen and the number of cells to be injected should be taken into account. The finer the needle, the greater the pressure exerted on the plunger and the more likely it is to cause lesions on the membrane of these cells to be transplanted or even burst. An important part of the cells can die at this stage without having been introduced into the muscle of the recipient. In this case, both cell debris, damaged cells and whole cells are introduced.
Puncture of the recipient’s muscle with the needle may damage some blood vessels and generate micro-hemorrhages around the transplanted cells. Hemorrhages then activate the mechanisms of vascular and plasma coagulation. It is highly likely that the transplanted cells can be trapped in a fibrin network. This eventuality can be the basis of hypoxia and the lack of nutritional intake that can lead to the death of transplanted cells. One study showed that migration of transplant cells could be improved by using a plasminogen activator, urokinase. Plasminogen converted by urokinase to plasmin can degrade the fibrin formed during coagulation as well as the extracellular matrix of the muscle fibers [41].
One of the mechanisms of cell death that has not yet been studied even partially is autophagy [42]. This death of a number of cells permits the survival of other cells. It is a form of cannibalism at the cellular level. Indeed, the macromolecules of the dead cells following autophagy are degraded by the autophagolysosomal machinery into small molecules that can be assimilated by the surviving cells. All transplantation trials indicated the survival of a small core of transplanted cells that differentiate and fuse with the host muscle fibers [38]. Are these cells that escaped autophagy? Demonstration of enhanced expression of autophagic markers in the micro-environment of the graft may be evidence of activation of this cell death pathway. It can also confirm the lack of nutrients in the microenvironment of the graft.
The Advances and Challenges of Gene Therapy
Before discussing gene therapy, we think a brief reminder of the DMD gene and the protein it encodes, dystrophin, is required for a better understanding of the different gene therapy approaches. The DMD gene is located on the short arm of the X chromosome in the locus 21 (Xp21). It is the largest gene in the human body with 2.4 million base pairs (2.4 Mb). It contains 79 exons (coding sequences) and its complementary DNA (cDNA) is 14 kilobases (14 kb) [5].
The protein encoded by the DMD gene is dystrophin. It is a long filament formed by 3685 amino acids with a molecular weight of 427 kDa. The secondary structure shows that dystrophin consists of 24 spectrin-like repeats (SLRs) and 4 junction regions called hinges (H1, H2, H3 and H4) [43]. Each SLR comprises three alpha helices: helix A, helix B and helix C (Figure 2a and 2b) [44]. Dystrophin allows the junction between a group of proteins in the sarcolemma (the dystrophin associated protein complex, DAPC)) and some cytosol proteins. The dystrophin complex plays a crucial role in maintaining the integrity of sarcolemma during muscle contractions [45]. Dystrophin is a shock absorber at the level of the muscle fiber and its absence, following some mutations, leads to the rupture of the sarcolemma and local necrosis of the muscle cell. 70% of DMD mutations are deletions of one or several exons that lead to a reading frame shift. The other 30% mutations are ponctual mutations that lead to a frame shift or the formation of a stop codon. Most of the deletions occur in the region between exons 44 and 56. This region is considered the hot region of the DMD gene [46,47]. The different approaches of gene therapy that we will describe in the following sections, concern the correction of the DMD gene in this region (Figure 2A,B) [48,49].
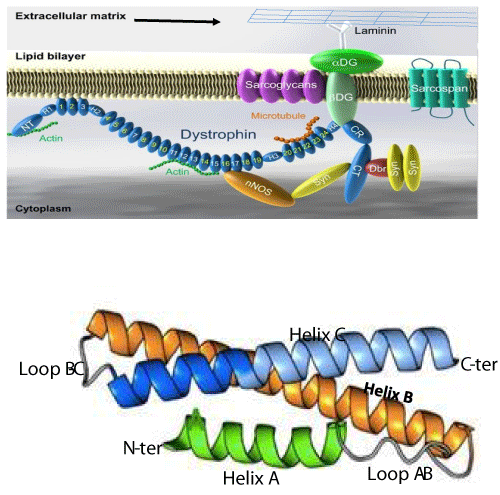
Figure 2: Dystrophin complex. A) Dystrophin contains an N-terminus (NT), a filament-shaped central part, a cysteine-rich (CR) domain and a C-terminal (CT). The central part is composed of 24 Spectrin-Like Repeats (SLRs) numbered from 1 to 24 and four junction regions called Hinge numbered H1 to H4. There are two actin binding sites, one of which is located at the NT end and the other at the SLR11-15. SLR1-3 interact with the cell membrane and SLR 16 and 17 with neuronal Nitric Oxide Synthetase (nNOS). Dystrophin also interacts with microtubules at SLR20-23. A portion of H4 and the CR domain bind with the beta-dystroglycan (βDG) protein. CT interacts with syntrophin (Syn) and dystrobrevin (Dbr). Dystrophin complex allows the binding of cytoskeletal proteins (actin and microtubules) with laminin in the extracellular matrix (arrow). Sarcoglycans and sarcospan do not interact directly with dystrophin. However, they stabilize the dystrophin complex. The spectral type repeat α-helices. Image modified from [48]. B) Dystrophin comprises 24 SLRs. Each SLR is formed by 3 alpha helices (helixes A, B and C). The N-terminus (N-ter) is at the beginning of helix A and the C-terminus (C-ter) is at the end of helix C. The helix A is bonded to helix B by loop AB while the helix B is connected to helix C loop BC. Image modified from [49].
Gene therapy aims to develop safe and systemic therapeutic approaches capable of restoring dystrophin expression in all skeletal muscles and heart of Duchenne patients [50]. Given the difficulty in introducing the complete DMD gene into the patient’s muscle fibers due to its large size, delivery of a truncated DMD gene or modifications of the existing DMD gene are being investigated to restore the reading frame either by exon skipping, by exonic knock-out or by the additional deletions of exon fragments. The ultimate aim of these approaches is to produce an internally truncated functional dystrophin protein to transform the Duchenne patient with a severe phenotype into a Becker type patient with a mild phenotype [51]. The idea of producing an internally truncated dystrophin is based on an observation made in a Becker patient who had a 46% deletion of the DMD gene and who lived for about 60 years, walking with a cane [52]. There are also other approaches to introduce a truncated DMD gene that codes either for a mini- or a micro-dystrophin using viral vectors [53,54]. Other approaches aim to suppress a premature stop codon [55].
The advances and challenges of exon skipping
Exon skipping is a gene therapy approach which consists in administering systemic or intramuscular RNA or DNA antisense oligonucleotides (ASO) to the animal model or the patient to mask the splice sites of the exonic sequences to delete them from the final mRNA [56]. With this approach, it is possible to eliminate one or several exons to restore the reading frame and thus allow the translation of an mRNA deleted of a part of its sequence. This permits the expression an internally truncated dystrophin protein which may be more or less functional.
The first experiments of this approach were performed in the mdx mice (a mouse model with a stop codon in exon 23 of the DMD gene) [57] and in myoblasts of patients [58]. Two proof-of-concept clinical trials were conducted by intramuscularly administering 2’-O-methyl-ribo-oligonucleotide-phosphorothioate, Drisapersen (PRO051/GSK402968, Prosensa / Biomarin) [10] and by IV administering phosphorodiamidate morpholino oligomer (PMO) Eteplirsen (AVI 4658/Exondys 51 Sarepta Therapeutics) [59,60] in the patient. These two ASOs generated a specific exon jump of exon 51 and induced the synthesis of the variable amounts of truncated dystrophin.
In case of Drisapersen (GSK402968), the results of the phase III trials did not bring significant improvements of the 6-minute walk test [61]. Several causes may explain these disappointing results: the lack of effect of ASOs in the cardiac muscle, the low penetration of ASOs into the skeletal muscle fibers, and the rapid elimination of ASOs from the circulation, which required repeated administration [50].
This situation led the FDA and the European Union to suspend the treatment of Duchenne muscular dystrophy with Drisapersen. To address this situation, modifications have been made to the exon skip to improve ASO activity. Some authors have used small nuclear RNAs (snRNAs), such as U7snRNAs, inserted into scAAV9 instead of ASOs to generate exon jump [62].
To improve the penetration of PMOs (phosphorodiamidate morpholino oligomers) into muscle fibers, some studies have used CPP (cell penetrating peptide) in complex with the PMOs [59]. To improve bio-distribution, a new class of ASOs has emerged. These are tricyclo-DNA (tcDNA) oligomers. This type of ASO can easily penetrate the skeletal muscles, the heart and the brain [63].
It is in this context that Sarepta Therapeutics initiated Phase I clinical trials in 2014 with PMOs, Eteplirsen (Exondys 51) for skipping of exon 51 that can correct the DMD gene in 13% of Duchenne patients. That clinical trial should have continue until 2019. But unexpectedly, the FDA authorized the commercialization of Eteplirsen as a treatment for DMD. More surprising is the statement made by Mr. Janet Woodcock, director of FDA Center for Drug Evaluation and Research: “Accelerated approval makes this drug available to patients based on initial data, but we eagerly await learning more about the efficacy of this drug through a confirmatory clinical trial that the company must conduct after approval » [64]. This unprecedented and controversial approval was motivated, according to some researchers, by the financial interests and was not based on a scientific basis proving the effectiveness of the drug.
Finally, some authors have proposed to skip multiple exons by administering a mixture of the several ASOs which must target several sequences of the pre-mRNA. Theoretically, it is possible to delete sequences ranging from exon 45 to exon 55 and to hope to correct the DMD gene in 63% of Duchenne patients [65]. However this proposed therapeutic approach does not take into account that exons 42-45 (SLR17) code for a part of the dystrophin protein which binds to nNOS [49]. The absence of nNOS leads to a more severe dystrophy [66-68].
Despite the advances observed in the treatment of DMD by exon skipping, this therapeutic approach has a number of disadvantages. This treatment requires a lifelong administartion of the ASOs because target the mRNA rather than the DMD gene. It is currently difficult to predict the long-term side effects of using ASOs.
Advances and challenges of recombinant endonucleases
In the introduction, we distinguished two types of recombinant endonucleases used for genome editing. These are the endonucleases, which recognize the target sequence by means of a polypeptide chain [12-14], and those, which recognize the target sequence by means of an RNA-type polynucleotide sequence [69]. Both types were used for correction of the dystrophin gene in the myoblasts of Duchenne patients and in different animal models using viral and plasmid vectors [70-73].
Advances and challenges of using meganucleases (MGNs)
Meganucleases are endonucleases that recognize long DNA sequences (14-40 bp) and generate DSBs. They may act as monomers or as homodimers [74-76]. Meganucleases were used to re-establish the reading frame in the Duchenne dystrophic patient’s myoblasts by generating DSBs and micro-insertions or micro-deletions (INDELs) [77]. They were also used in the immortalized myoblasts of the patient to generate DSBs and allow the insertion of exons 45-52. The sequences of these exons (4.5 kb cDNA) were inserted into a donor lentivirus and integrated into the DMD mutated gene of the patient’s myoblasts thus allowing synthesis of a normal dystrophin [71]. Despite its high precision, the production of recombinant meganucleases that can recognize specific DNA sequences remains a challenge and therefore limits their use.
Advances and challenges of Zinc Finger Nucleases (ZFNs)
Zinc Finger Nucleases are hybrid proteins produced from a chimeric gene formed by the fusion of the gene encoding the Zinc Finger Protein (ZFP) [78], on the one hand and the gene encoding the catalytic region of the restriction enzyme FokI on the other hand [13]. ZFPs belong to the Cys2-His2 family of zinc finger proteins. Each zinc finger consists of 25 amino acids of which 10 form the antiparallel β sheet (amino acids 1 to 10) and 12 the α-helix (amino acids 12 to 24). Four amino acids (His 19, His 23, Cys 3 and Cys 6) bind the zinc atom to the α-helix and the β-sheet respectively and stabilize the ZFP structure. The interaction with the DNA takes place at the level of the α-helix. The restriction enzyme FokI is a protein of 587 amino acids produced by the bacterium Flavobacterium okeanokoites. It includes a DNA-binding N-terminus and a catalytic C-terminus [79]. ZFNs act as the left (G) and right (D) antiparallel dimers separated from 5 to 7 nucleotides when linked to DNA [80-82]. Each monomer comprises 3 to 6 zinc finger and each zinc finger recognizes 3 nucleotides. The catalytic portion of FokI is attached to the C-terminus of each monomer. ZFNs were used to correct the mutated DMD gene in mice [83] and in Duchenne cells in culture by excision of exon 51 of the DMD gene [72]. Theoretically, excision of exon 51 can allow the correction of DMD gene in 13% of Duchenne patients. The biggest challenge for the use of ZFNs in gene therapy is the great difficulty in their production.
Advances and challenges of Transcription Activator Like-Effector Nucleases (TALENs)
TALENs are hybrid recombinant proteins produced from a chimeric gene derived from the fusion of the gene encoding the central domain of TALE (Transcription Activator Like-Effector) on one hand and that encoding the catalytic part of FokI [84]. TALEs are proteins produced by Xanthomonas bacteria that infect plants and act as transcription factors. These proteins consist of an N-terminal end with a translocation domain (TD), a DNA binding binding domain and a C-terminal end comprising a localization sequence (NLS) and a transcriptional activation domain (TAD). DNA binding is achieved through the TALE portion. The FokI nuclease is use to induce cleavage. The repeated central domain is formed from 15.5 to 19.5 monomers and each monomer consists of 33 or 34 highly conserved amino acids. The last monomer is composed of only 20 amino acids and is considered as half-monomer. The binding specificity of each monomer with DNA is essentially defined by the amino acid polymorphism at position 12 and 13 (Repeat-Variable Di-residues, RVDs). Each monomer recognizes a nucleotide by the two amino acids located at positions 12 and 13. Thus, the following code is used: HD for attachment to cytosine, NI for adenine, NG for thymine and NN for guanine [85-87]. The repeated central domain can specifically recognize up to 15 nucleotides. Like the ZFNs, TALENS also act as dimers [88]. The monomer G and the monomer D are antiparallel and have, at their C-terminus, the catalytic part of FokI. TALENs corrected the mutated dystrophin gene in myoblasts and fibroblasts [89] and in patient induced pluripotent cells [73]. Like the ZFNs, the challenge of using TALENs as a therapeutic approach against DMD lies in their construction.
Advances and challenges of using the CRISPR/Cas9
The Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR) system, CRISPR/associated 9 (Cas9) is a defense mechanism used by bacteria to destroy phagic and plasmid DNA [90]. The type most commonly used is type II of Streptococcus pyogenes. When used for genome editing in eukaryotic cells, type II comprises a single guide RNA (sgRNA) sequence formed of two parts: a variable portion of 20 nucleotides, which recognizes the target DNA sequence, and a conserved portion of 110 nucleotides, which stabilizes the binding of the Cas9 nuclease at the cleavage site. For SpCas9 to be recruited on the target sequence, this sequence has to be followed in 3’ by an NGG trinucleotide called Protospacer adjacent motif (PAM) (Figure 3). There are two catalytic sites in Cas9: HNH and RuvC. DSBs are generated exactly at 3 nucleotides from the PAM toward the 5 ‘end of the target DNA [91-95].
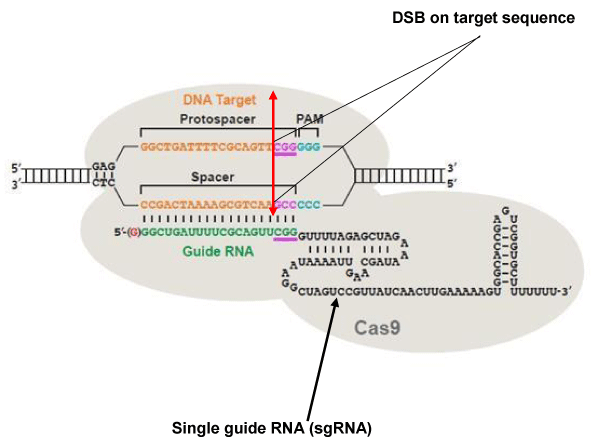
Figure 3: The CRISPR / Cas9 system. The DNA target sequence (orange) is illustrated with the two strands separated. The sense strand of target sequence id followed at its 3’ end by the protospacer adjacent motif (PAM), which is NGG (in blue) for the SpCas9 nuclease. The cleavage site (double arrow in red) is located at 3 nucleotides (CGG in mauve) from the PAM towards the 5 ‘end. The variable portion of sgRNA (20 nucleotides in green) recognizes the target sequence by the Watson-Crick base pairing. The nuclease Cas9 (in light gray) is recruited at the PAM. The invariable part of sgRNA (nucleotides in black) stabilizes the whole structure. Image modified from [107].
The main advantage of the CRISPR/Cas9 system over the other endonucleases described above is its production simplicity. The only limiting factor in the targeting of DNA sequences is the presence of the NGG PAM required for the SpCas9 nuclease. However, in recent years, other CRISPR systems (Cpf1, SaCas9, CjCas9...) [96-98] with different PAMs have been identified in many bacterial species and are being tested, thus increasing our ability to target virtually the entire genome. In addition to ease of production, the CRISPR/Cas9 system has demonstrated high efficiency and accuracy when used in vitro and in vivo in eukaryotic cells and in animal models when inserted into plasmids and viral vectors [99,100]. For Duchenne muscular dystrophy gene therapy, several studies using the CRISPR/Cas9 system have already been published. Some studies aim to produce an internally truncated dystrophin that could be functional by generating intronic DSBs followed by deletions of complete exons to restore the reading frame shift, which was responsible for the presence of a premature stop codon [101-103]. The problem with this approach is that it does not take into account the structure of truncated dystrophin. To function optimally, the structure of the SLR of the truncated dystrophin produced must be correct. Each SLR must be made of a succession of 3 alpha helixes: A, B and C. A disturbance in this arrangement results in poor conformation of the truncated dystrophin and hence in a less optimally functional truncated protein. This explains the existence of numerous Becker phenotypes, some of which (with truncated dystrophins with poor conformation) are severe [104]. It is therefore not enough to restore the reading frame only to produce an internally truncated dystrophin. It is for this reason that we have used the CRISPR-induced deletion method (CinDel) [105,106]. CinDel aims to generate DSBs with two sgRNA and the Cas9 nuclease in the exons preceding and following the patient deletion, to induce additional deletions of portions of the target exons and of intron sequences located between these two DSBs. This generates a hybrid exon, which not only restores the reading frame but also allows the expression of an internally truncated dystrophin. The judicious choice of sgRNAs makes it possible to produce a dystrophin whose structure respects the order of succession of the helices in the SLRs, which may have a good conformation and which will functional optimally. The in vitro tests on the myoblasts of the patient and in vivo on the animal model confirmed the synthesis of an internally truncated dystrophin. Some other authors aim to generate DSBs and insert a short cDNA fragment using a viral vector. By homologous recombination, the dystrophin gene can be corrected and normal dystrophin produced [99]. The CRISPR/Cas9 system has revolutionized the gene therapy approach and to date, numerous studies are being conducted around the world to effectively treat a large number of monogenic genetic diseases. The major challenge in using this approach is the possibility of off-target mutations (Figure 3) [107].
Replacing the DMD mutated gene with a mini-dystrophin synthetic gene
In 2006, Asklepios Biopharmaceuticas Inc. undertook a Phase I clinical trial of its product BiostrophinR, a mini-dystrophin gene sequence consisting of the N-terminal domain (NTD), hinge 1 (H1), SLR1-3), hinge 3 (H3), SLR20-24), hinge 4 (H4), a cysteine rich domain (CRD) and the C terminal domain. In summary, this is a mini-gene having this structure: NTD-H1-R1-R2-R3-H3-R20-R-21-R22-R23-R24-H4-CRD-NTD. It has a size of about 4.5 Kb and was inserted into an AAV2.5 under a CMV promoter. The product was administered intra-muscularly to 6 patients [108]. The test did not improve the condition of the patients and an immune reaction was observed against the transgene and the viral vector [9].
Suppression of the nonsense mutations
About 10% of DMD patients have a nonsense point mutation in the dystrophin gene. This type of mutation leads to premature termination of dystrophin mRNA translation and ultimately to the absence of dystrophin in the patient’s muscle fibers [47]. PTC-Therapeutics Inc. has developed the AtularenR product (PTC-124), a drug, which permits to bypass the premature stop codon and allowing the synthesis of dystrophin in some dystrophic patients. A phase III clinical trial was performed in 174 patients [109]. The results of this trial indicated that this approach could be used to treat some DMD patients [110]. However, the lifetime use of this drug may have undesirable effects.
The model animals of muscular dystrophy
Approaches of gene therapy for DMD invariably follows the following pathway: in vitro test in patient cells or in vivo tests in animal models, clinical trials Phase I, Phase II and Phase III. To date, there are several animal models used to better understand the mechanisms governing the disease and used for preclinical trials [48]. Each model has advantages and disadvantages. In the next section, we will examine some models that are commonly used for DMD.
Mouse mdx
The mdx mouse contains a nonsense point mutation (a C to T transition) in exon 23 of dystrophin gene [111]. This type of mutation, as mentioned above, represents only about 10% of cases in humans [47]. The use of immortalized DMD cells in vitro does not pose a problem with regard to the type of mutation, however, the use of a model, which has a different phenotype and a different mutation, is not the best situation. The vast majority of preclinical in vivo assays have been performed in the mdx mouse animal model. Compared to the DMD phenotype, the phenotype of the mdx mouse is far less severe. There are many reasons for this difference. First in mdx mice, there is an over-expression of utrophin, a cytosolic protein that has a structure similar to that of dystrophin, which may compensate for the absence of dystrophin [112]. The second reason is the almost continuous renewal of mouse satellite cells (myogenic progenitor cells), which provide a sustained repair of damaged muscle fibers [113]. The third reason is the presence of the cytidine monophosphate sialic acid hydroxylase (CMHA) gene in mdx mice, whereas this gene is repressed in humans [114]. The fourth reason is the small size of the model and therefore its low muscle mass [115]. For all these reasons, some authors claim that the mdx model is not the best and that model results are not often transposable to humans [19].
Instead of mdx mice, it would be better to use hDMD/mdx mice [116]. It is an mdx mouse in which the complete (2.4 Mb) DMD gene has been introduced into the zygote by fusing it with a Yeast Artificial Chromosome (YAC) spheroblast containing the transgene. The human transgene located on chromosome 5 of the mdx mouse can be mutated (our team is already working with a view to producing hDMD/mdx mice with the transgene deleted of some exons using the CRISPR/Cas9 system to reproduce the same mutations as in humans). This new model could then be corrected with the therapy approaches described above. This model has the advantage that the corrections are made on the human gene using the same sgRNAs as used for the DMD patient myoblasts. Thus the results can be, to some extent, transposed to humans. Thus the sgRNA/Cas9, ZFN and TALEN showing activity on the transgene, will also be active on the same gene in humans.
Dystrophic Dogs
The earliest descriptions of dystrophic dogs date back more than 50 years [117]. Among the different models, Golden Retriever Muscular Dystrophy (GRMD) [118,119] and Cavalier King Charles Spaniel Muscular Dystrophy (CKCS-MD) are the best known [120]. GRMD is the result of a mutation at the splice site at intron 6 (5 ‘splicer donor). This mutation leads to the exon 7 skipping during the maturation of dystrophin mRNA, this causes a premature termination of translation and the absence of dystrophin in the muscle fibers of the dog. In the case of CKCS-MD, this is also a mutation at the splice site in intron 50 (5 ‘splicer donor). This mutation leads to the skipping of exon 50 in the dystrophin mRNA and to a reading frame shift leading to the absence of dystrophin. The canine models have a more severe phenotype and are close to the human phenotype than the mdx mouse model. Unfortunately, this model is very expensive and also difficult to reproduce.
Dystrophic Rats
Dystrophic rats were produced by deleting exons 3-16 of the rat DMD gene with two sgRNA/Cas9. The sgRNAs and the Cas9 mRNA were injected into the rat zygotes generating the deletion of the target exons. The joining of the exons 2 and 17 generated a reading frame shift preventing the synthesis of complete dystrophin protein. Besides the increase in size (about 10 times the size of the mdx mouse), mdx rats exhibit a more severe disease phenotype. Hypertrophy of certain skeletal and cardiac muscles is observed [121]. Another model of dystrophic rat produced with TALENs by deleting exon 23, also showed a severe phenotype as the animal advanced in age. The presence of fibrosis and the infiltration of adipose tissue in certain skeletal muscles has been noted [122]. This is model, which is not too expensive, that can be used to replace the mdx mouse.
Dystrophic rhesus macaque
To produce the dystrophic macaques, exons 4 and 46 belonging to two respective hot-spot regions of the DMD gene (exon region 3-7 and exons 44-56 region) were targeted. Both sgRNAs and Cas9 mRNA were injected into rhesus macaque zygotes. These sgRNA/Cas9 generated DSBs and INDELs at the target sites. Some of these INDELs induced a reading frame shift or premature stop codons into the mutated gene [123]. The use of a non-human primate model despite its high cost could be a crucial step before clinical trials.
Dystrophic Pig
The dystrophic pig model was produced to effectively solve the problem of low muscle mass in the vast majority of animal models used. This model also showed a phenotype as severe as the human phenotype of DMD [124].
CONCLUSION AND FUTURE PROSPECTS
Cell and gene therapy of muscular dystrophies has benefited from considerable advances in fields as diverse as synthetic biology, genomics, epigenetics… The application of these advances in gene therapy has allowed a good understanding of different therapeutic approaches. The wide progresses of recombinant endonucleases, particularly the CRISPR/Cas9 system, permit not only in the development of gene editing therapy, but also the production of new animal models. This augurs well in a near future for curative treatments of genetic diseases in general and Duchenne muscular dystrophy in particular.
ACKNOWLEDGMENT
This work has been supported by grants from the Foundation from Cell and gene Therapy and from the Canadian Institute of Health Research (grant # 271774).
REFERENCES
- Consortium, IHGS (2004). Nature 431: 931-945.
- Consortium, TEP (2012). Nature 489: 45-77.
- Kunkel L, Hejtmancik J, Caskey C, Speer A, Monaco AP, et al. Analysis of deletions in DNA from patients with Becker and Duchenne muscular dystrophy. Nature. 1986; 322: 73-77. Ref.: https://goo.gl/MtkNij
- Bodrug SE, Ray PN, Gonzalez IL, Schmickel RD, Sylvester JE, et al. Molecular analysis of à constitutional X-autosome translocation in a female with muscular dystrophy. Science.1987; 237: 1620-1624. Ref.: https://goo.gl/XC8UBR
- Hoffman EP, Brown RH Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987; 51: 919-928. Ref.: https://goo.gl/Mvt6Xz
- Hoffman EP, Fischbeck KH, Brown RH, Johnson M, Medori R, et al. Characterization of dystrophin in muscle biopsy specimens from patients with Duchenne’s or Becker’s muscular dystrophy. N Engl J Med.1988; 318: 1363-1368. Ref.: https://goo.gl/iACqAb
- Partridge TA, Grounds M, Sloper JC. Evidence of fusion between host and donor myoblasts in skeletal muscle grafts. Nature. 1978; 273: 306-308. Ref.: https://goo.gl/eBeWSs
- Skuk D. Myoblast transplantation for inherited myopathies: a clinical approach. Expert Opin Biol Ther. 2004; 4: 1871-1885. Ref.: https://goo.gl/g7ct6W
- Mendell JR, Campbell K, Rodino‐Klapac L, Sahenk Z, Shilling C, et al. Dystrophin immunity in Duchenne's muscular dystrophy. N Engl J Med. 2010; 363: 1429-1437. Ref.: https://goo.gl/E1YG6t
- van Deutekom, Janson AA, Ginjaar IB, Frankhuizen WS, Aartsma-Rus A, et al. Local dystrophin restoration with antisens oligonucléotides PRO051. N Engl J Med. 2007; 357 : 2677-2686. Ref.: https://goo.gl/o9btPx
- Voit T, Topaloglu H, Straub V, Muntoni F, Deconinck N, et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo‐controlled phase 2 study. Lancet Neurol. 2014; 13: 987-996. Ref.: https://goo.gl/kJAv3X
- Gouble A, Smith J, Bruneau S, Perez C, Guyot V, et al. Efficient in toto targeted recombination in mouse liver by meganuclease-induced double-strand break. J Gene Med. 2006; 8: 616-622. Ref.: https://goo.gl/fv4nKV
- Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A. 1996; 93: 1156-1160. Ref.: https://goo.gl/adR8MB
- Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nature Reviews Molecular Cell Biology. 2013; 14: 49-55. Ref.: https://goo.gl/azsxQ4
- Jinek M, Chvlinski K, Fonfara I, Hauer M, Doudna JA, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337: 816-821. Ref.: https://goo.gl/CmHA49
- Watt DJ, Lambert K, Morgan JE. Incorporation of donor muscle precursor cell into an area of muscle generation in the host mouse. J neurol Sci. 1982; 57: 319-331.
- Lipton BH, Schultz E. Developmental faite of skeletal muscle satellite cells. Science. 1979; 205: 1292-1294. Ref.: https://goo.gl/jy1GiZ
- Yao SN, Kurachi K. Implanted myoblasts not fuse with myofibers but also survive as muscle precursor cells. J Cell Sci. 1993; 105: 957-963. Ref.: https://goo.gl/8DDr6R
- Skuk D. Cell Transplantation and ‘‘Stem Cell Therapy’’ in the treatment of myopathies: many promises in mice, few realities in humans. Hindawi Publishing Corporation ISRN Transplantation. 2011; 25.
- Zheng B, Cao B, Crisan M, Sun B, Li G, et al. Prospective identification of myogenic endothelial cells in human skeletal muscle. Nature Biotechnology. 2007; 25: 1025-1034. Ref.: https://goo.gl/u1gywA
- Sampaolesi M, Blot S, D’Antona G, Granger M, Tonlorenzi R, et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature. 2013; 494. Ref.: https://goo.gl/z9h9Hy
- Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008; 3: 301-313. Ref.: https://goo.gl/FJVRMZ
- Benchaouir R, Meregalli M, Farini A, D’Antona G, Belicchi M, et al. Restoration of human dystrophin following transplantation of exon-skipping engineered DMD patient stem cells into dystrophic mice. Cell Stem Cell. 2007; 1: 646-657. Ref.: https://goo.gl/uRsuCx
- Vauchez K, Marolleau J, Schmid M, Khattar P, Chapel A, et al. Aldehyde dehydrogenase activity identifies a population of human skeletal muscle cells with high myogenic capacities. Molecular therapy, 2009; 17: 1948-1958. Ref.: https://goo.gl/td98pa
- Mitchell KJ, Pannerec A, Cabot B, Parlakian A, Besson V, et al. Identification and characterization of à non-satellite cell muscle resident progenitor during postnatal development. Nature Cell Biology. 2010; 12: 257-266. Ref.: https://goo.gl/BjVqCZ
- Liadaki K, Casar JC, WMe. Beta-4-integrin marks interstitial myogenic progenitor cells in adult murine skeletal muscle, Journal of Histochemistry and Cytochemistry. 2012; 60: 31-44.
- Zhao C, Farruggio AP, Bjornson CRR, Chavez CL, Geisinger JM, et al. Recombinase-Mediated Reprogramming and Dystrophin Gene Addition in mdx Mouse Induced Pluripotent Stem Cells. PLoS One. 2014; 9 : e96279. Ref.: https://goo.gl/zPS8gz
- Skuk D, Goulet M, Roy B, Chapdelaine P, Bouchard JP, et al. Dystrophin expression in muscles of Duchenne muscular patients after high-density injection of normal myogenic cells. J Neuropathol Exp Neurol. 2006; 65: 371-386. Ref.: https://goo.gl/iBPfD2
- Lazerges C, Daussin PA, Coulet B, Boudbaker el Andalousi R, Micallef JP, et al. Transplantation of primary satellite cells improves properties of reinnervated skeletal muscles. Muscle Nerve. 2004; 29: 218-226. Ref.: https://goo.gl/W3VRyf
- Skuk D, Tremblay JP. Intramuscular cell transplantation as a potential treatment of myopathies: clinical and preclinical relevant data. Expert Opin Biol Ther. 2011; 11: 359-374. Ref.: https://goo.gl/3bxS1u
- Irintchev A, Langer M, Zweyer M, Theisen R, Wemig A. Functional improvement of damaged adult mouse muscle by implantation of primary myoblasts. J physiol. 1997; 1500: 775-785. Ref.: https://goo.gl/u5wJsw
- Wernig A, Zweyer M, Irintchev A. Function of skeletal muscle tissue formed after transplantation indo irradiated mouse muscles. J Physiol. 2000; 522: 333-345. Ref.: https://goo.gl/7RkSwF
- Torrente Y, Belicchi M, Sampaolesi M, Pisati F, Meregalli M, et al. Human circulating AC133+ stem cells restore dystrophin expression and ameliorate function in dystrophic skeletal muscle, Journal of Clinical Investigation. 2004; 114: 182-195. Ref.: https://goo.gl/WQpNGc
- Hardiman O, Sklar RM, Brown HR Jr. Direct effects of cyclosporin A and cyclophosphamide on differentiation of normal human myoblasts in culture. Neurology. 1993; 43: 1432-1434. Ref.: https://goo.gl/dQY6U7
- Kinoshita I, Vilquin JT, Guerette B, Asselin I, Roy R, et al. Very efficient myoblast allotransplantation in mice under FK506 immunosuppression. Muscle & Nerve. 1994; 17: 1407-1415. Ref.: https://goo.gl/swTnwK
- Chandrasekharan D, Issa F, Wood KJ. Achieving operational tolerance in transplantation: how can lessons from the clinic inform research directions?. Transplant International. 2013; 26 : 576-589. Ref.: https://goo.gl/jWEoxX
- Arpke, Darabi R, Mader TL, Zhang Y, Toyama A, et al. A new immuno- dystrophin-deficient model, the NSG-mdx(4Cv) mouse, provides evidence for functional improvement following allogeneic satellite cell transplantation. Stem cells. 2013; 31: 1611-1620. Ref.: https://goo.gl/2SrqUy
- Fukada S, Miyagoe-Suzuki Y, Tsukihara H, Yausa K, Higuchi S, et al. Muscle regeneration by reconstitution with bone marrow or fetal liver cells from green fluorescent protein-gene transgenic mice. J Cell Sci. 2002; 115: 1285-1293. Ref.: https://goo.gl/dJ1Yn3
- Partridge TA, Morgan JE, Coulton GR, Hoffman EP, Kunkel LM. Conversion of mdx myofibres from dystrophin-negative to -positive by injection of normal myoblasts. Nature. 1989; 337: 176-179. Ref.: https://goo.gl/BNw5ic
- Traggiai E, Chicha L, Mazzucchelli L, Bronz L, Piffaretti JC, et al. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science. 2004; 304: 104-107. Ref.: https://goo.gl/yYFWMx
- El Fahime E, Mills P, Lafreniere JF, Torrente Y, Tremblay JP. The urokinase plasminogen activator: an interesting way to improve myoblast migration following their transplantation. Exp Cell Res. 2002; 280: 169-178. Ref.: https://goo.gl/nxGa13
- Ferraro E, Cecconi F. Autophagic and apoptic response to stress signals in mammalian cells. Archives of Biochemistry and Biophysics. 2007; 462: 210-219. Ref.: https://goo.gl/QqBjpc
- Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988; 53: 219-228. Ref.: https://goo.gl/cG9dnz
- Cohen C, Parry A D. a-helical coiled coils; à widespread motif in proteins. Trends Biochem Sci. 1986; 11: 245-248.
- AE, E (1990). Dystrophin function. Lancet 335: 1289.
- Outersrout DG, Kabadi AM, Majoros WH, Reddy TE, Thakore PI, et al. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat Commun. 2015; 6:6244. Ref.: https://goo.gl/wRi4wk
- Bladen CL, Salgado D, Monges S, Foncuberta ME, Kekou K, et al. The TREAT‐NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015; 36: 395-402. Ref.: https://goo.gl/1BTyPF
- McGreevy JW, Hakim CH, McIntosh MA, Duan D. Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis Model Mech. 2015; 8: 195-213. Ref.: https://goo.gl/Ric8Wq
- Lai Y, Thomas GD, Yue Y, Yang HT, Li D, et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest. 2009; 119: 624-635. Ref.: https://goo.gl/5bvFdS
- Guiraud S, Chen H, Burns DT, Davies KE. Advances in genetic therapeutic strategies for Duchenne muscular dystrophy. Experimental Physiology. 2015; 100: 1458-1467. Ref.: https://goo.gl/329EDH
- Hoffman E. Genotype/phenotype correlations in Duchenne/Becker dystrophy. Mol Cell Biol Hum Dis Ser. 1993; 3: 12-36. Ref.: https://goo.gl/GR1aVE
- England SB, Nicholson LV, Johnson MA, Forrest SM, Love DR, et al. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature. 1990; 343: 180-182. Ref.: https://goo.gl/EsyThj
- Hayashita‐Kinoh H, Yugeta N, Okada H, Nitahara‐Kasahara Y, Chiyo T, et al. Intra‐amniotic rAAV‐mediated microdystrophin gene transfer improves canine X‐linked muscular dystrophy and may induce immune tolerance. Mol Ther. 2015; 23: 627-637. Ref.: https://goo.gl/ZeeCDD
- Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, et al. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med. 2002; 8: 253-261. Ref.: https://goo.gl/Aty4cb
- Malik V, Rodino‐Klapac LR, Viollet L, Wall C, King W, et al. Gentamicin‐induced readthrough of stop codons in Duchenne muscular dystrophy. Ann Neurol. 2010; 67: 771-780. Ref.: https://goo.gl/KjGncc
- Aartsma-Rus A, van Ommen GJ. Antisense-mediated exon skipping: A versatile tool with therapeutic and research applications. RNA. 2007; 13: 1609-1624. Ref.: https://goo.gl/THGt6o
- Dunkley MG, Manoharan M, Villiet P, Eperon IC, Dickson G. Modification of splicing in the dystrophin gene in cultured mdx muscle cells by antisenses oligoribonucleotides. Hum mol Genet. 1998; 7: 1083-1090. Ref.: https://goo.gl/KB9fSM
- van Deutekom JC, Bremmer‐Bout M, Janson AA, Ginjaar IB, Baas F, et al. Antisense‐induced exon skipping restores dystrophin expression in DMD patient derived muscle cells. Hum Mol Genet. 2001; 10: 1547-1554. Ref.: https://goo.gl/gJUPGS
- Kinali M, Arechavala‐Gomeza V, Feng L, Cirak S, Hunt D, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI‐4658 in Duchenne muscular dystrophy: a single‐blind, placebo‐controlled, dose‐escalation, proof‐of‐concept study. Lancet Neurol. 2009; 8: 918-928. Ref.: https://goo.gl/rMgxds
- Mendell JR, Rodino‐Klapac LR, Sahenk Z, Roush K, Bird L, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013; 74: 637-647. Ref.: https://goo.gl/TkUMi8
- http://www.gsk.com
- Goyenvalle A, Vulin A, Fougerousse F, Leturcq F, Kaplan JC, et al. Rescue of dystrophic muscle through U7 snRNA‐mediated exon skipping. Science. 2004; 306: 1796-1799. Ref.: https://goo.gl/WBj2PP
- Goyenvalle A, Griffith G, Babbs A, EI Andaloussi S, Ezzat K, et al. Functional correction in mouse models of muscular dystrophy using exon‐skipping tricyclo‐DNA oligomers. Nat Med. 2015; 21: 270-275. Ref.: https://goo.gl/wT3Dbe
- http://www.streetinsider.com/...Sarepta+Therapeutics+(SRPT)+Eteplirsen
- Béroud C, Tuffery‐Giraud S, Matsuo M, Hamroun D, Humbertclaude V, et al. Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Hum Mutat. 2007; 28: 196-202. Ref.: https://goo.gl/1burBb
- Stamler JS, Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev. 2001; 81: 209-237. Ref.: https://goo.gl/LtpbR8
- Bredt DS. Endogenous nitric oxide synthesis: biological functions and pathophysiology. Free Radic Res. 1999; 31: 577-596. Ref.: https://goo.gl/sZN9wb
- Heydemann, AaM E. NO more muscle fatigue. J Clin Invest. 2009; 119: 448-450.
- Jinek M, Chylinski K, Fonfara I, Hauer JA, Charpentier E. A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337: 816-821. Ref.: https://goo.gl/rwQsCi
- Jarmin S, Kymalainen H, Popplewell L, Dickson G. New development in the use of gene therapy to treat Duchenne muscular dystrophy. Expert Opin Biol Ther. 2014; 14: 209-230. Ref.: https://goo.gl/VaJhWC
- Popplewell L, Koo T, Leclerc X, Duclert A, Mamchaoui K, et al. Gene correction of a duchenne muscular dystrophy mutation by meganuclease-enhanced exon knock-in. Hum Gene Ther. 2013; 24: 692-701. Ref.: https://goo.gl/c9sTMY
- Ousterout D, Kabadi AM, Thakore PI, Perez-Pinera P, Brown M, et al. Correction of Dystrophin Expression in Cells From Duchenne Muscular Dystrophy Patients Through Genomic Excision of Exon 51 by Zinc Finger Nucleases. Mol Ther. 2015; 23: 523-532. Ref.: https://goo.gl/MzTxtF
- Li HL, Fujimoto N, Sasakawa N, Shirai S, Ohkame T, et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cell by TALENs and CRISPR-Cas9. Stem Cell Reports. 2015; 4: 134-154. Ref.: https://goo.gl/DpDtH7
- Chevalier B, Sussman D, Otis C, Noel AJ, Turmel M, et al. Metal-dependent DNA cleavage mechanism of the I-CreI LAGLIDADG homing endonuclease. Biochemistry. 2004; 43: 14015-14026. Ref.: https://goo.gl/FdVeL1
- Grizot S, Epinat JC, Thomas S, Duclert A, Rolland S, et al. Generation of redesigned homing endonucleases comprising DNA-binding domains derived from two different scaffolds. Nucleic Acids Res. 2010; 38: 2006-2018. Ref.: https://goo.gl/6NFHwf
- Silva G, Poirot L, Galetto R, Smith J, Montoya G, et al. Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Curr Gene Ther. 2011; 11: 11-27. Ref.: https://goo.gl/vM98zr
- Chapdelaine P, Pichavant C, Rousseau J, Pâques F, Tremblay JP. Meganucleases can restore the reading frame of a mutated dystrophin. Gene Ther. 2010; 17: 846-858. Ref.: https://goo.gl/QV9mrz
- Klug A. The discovery of Zinc Fingers and their applications in gene regulation and genome manipulation. Annu Rev Biochem. 2010; 79: 213-231. Ref.: https://goo.gl/MSdK7H
- Kim YG, Li L, Chandrasegaran S. Insertion and deletion mutants of FokI restriction endonuclease. J Biol Chem. 1994; 269: 31978-31982. Ref.: https://goo.gl/cixgTn
- Doyon Y, Vo TD, Mendel MC, Greenberg SG, Wang J, et al. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat Methods. 2011; 8: 74-79. Ref.: https://goo.gl/stXgXs
- Miller JC, Holmes MC, Wang J, Guschin DY, Lee YL, et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol. 2007; 25: 778-785. Ref.: https://goo.gl/i6fbF8
- Szczepek M, Brondani V, Buchel J, Serrano L, Segal DJ, et al. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol. 2007; 25: 786-793. Ref.: https://goo.gl/z4UxSJ
- Anguela XM, Sharma R, Doyon Y, Miller JC, Li H, et al. Robust ZFN-mediated genome editing in adule hemophilic mice. Blood.2013; 122: 3283-3287. Ref.: https://goo.gl/NZvMJe
- Mussolino C, Cathomen T. TALE nucleases: tailored genome engineering made easy. Curr Opin Biotechnol.2012; 23: 1-7. Ref.: https://goo.gl/ajCo7C
- Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009; 326: 1509-1512. Ref.: https://goo.gl/etRHCy
- Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009; 326: 1501. Ref.: https://goo.gl/iZLHkR
- Bogdanove AJ, Schornack S, Lahaye T. TAL effectors: finding plant genes for disease and defense. Curr Opin Plant Biol. 2010; 13: 394-401. Ref.: https://goo.gl/EpYj2E
- Hisano Y, Ota S, Arakawa K, Muraki M, Kono N, et al. Quantitative assay for TALEN activity at endogenous genomic loci. Biol Open. 2013; 2: 363-367. Ref.: https://goo.gl/VEi2Ev
- Ousterout DG, Perez-Pinera P, Thakore PI, Kabadi AM, Brown MT, et al. Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol Ther. 2013; 21: 1718-1726. Ref.: https://goo.gl/my1vk1
- Jinek M, East A, Cheng A, Lin S, Ma E, et al. RNA-programmed genome editing in human Cells. ELife. 2013; 2. Ref.: https://goo.gl/a964dZ
- Cong L, Ran FA, Cox D, Lin S, Barretto R, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013; 339: 819-823. Ref.: https://goo.gl/gQMiSy
- Doudna J, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014; 346: Ref.: https://goo.gl/Pg9LmR
- Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014; 32: 347-355. Ref.: https://goo.gl/26DN9d
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, et al. RNA-guided human genome engineering via Cas9. Science. 2013; 339: 823-826. Ref.: https://goo.gl/1nBQCh
- Cho S, Kim S, Kim JK, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013; 31: 230-232. Ref.: https://goo.gl/uSPnNX
- Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell. 2015; 163: 1-13. Ref.: https://goo.gl/SJmEB3
- Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015; 520: 186-191. Ref.: https://goo.gl/t3uR9f
- Kim E, Koo T, Park SW, Kim D, Kim K, et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat Commun. 2017; 8: Ref.: https://goo.gl/osgJzU
- Zheng Q, Cai X, Tan M, Schaffert S, Arnold C, et al. Precise gene deletion and replacement using the CRISPR/Cas9 system in human cells. Biotechniques. 2014; 57: 115-124. Ref.: https://goo.gl/LgZzML
- Bengtsson NE, Hall JK, Odom GL, Phelps MP, Andrus CR, et al. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat Commun. 2017; 8: Ref.: https://goo.gl/VbqZeq
- Tabebordbar M, Zhu K, Cheng JKW, Chew WL, Widrick JJ, et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2015; 35: 407-411. Ref.: https://goo.gl/qLqCG8
- Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, et al. In vivo genome editing imporves muscle function in a model of Duchenne muscular dystrophy. Science.2015; 350: 403-407. Ref.: https://goo.gl/8z98NS
- Long C, Amoasii L, Mireault AA, McAnally JR, Li H, et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016; 351: 400-403. Ref.: https://goo.gl/DpbqgY
- Nicolas A, Raguénès-Nicol C, Ben Yaou R, Ameziane-Le Hir S, Chéron A. Becker muscular dystrophy severity is linked to the structure of dystrophin. Hum Mol Genet. 2015; 24: 1267-1279. Ref.: https://goo.gl/vgcWbq
- Iyombe-Engembe JP, Ouellet DL, Barbeau X, Rousseau J, Chapdelaine P, et al. Efficient restoration of the dystrophin gene reading frame and protein structure in DMD myoblasts using the CinDel method. Mol Ther Nucleic Acids. 2016; 5: e283. Ref.: https://goo.gl/dzBUqc
- Tremblay JP, Iyombe-Engembe JP, Duchêne B, Ouellet DL. Gene Editing for Duchenne Muscular Dystrophy Using the CRISPR/Cas9 Technology: The Importance of Fine-tuning the Approach. Mol Ther. 2016; 24: 1888-1889. Ref.: https://goo.gl/5qTD6i
- Farboud B, Meyer BJ. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics. 2015; 199: 959-971. Ref.: https://goo.gl/qPtn3i
- http://www.askbio.comm
- http://www.ptcbio.com
- Bushby K, Finkel R, Wong B, Barohn R, Campbell C, et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve.2014; 50: 477-487. Ref.: https://goo.gl/96tuKV
- Bulfield C, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA. 1984; 1: 1189-1192. Ref.: https://goo.gl/LFz1gt
- Matsumura K, Ervasti JM, Ohlendieck K, Kahl SD, Campbell KP. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature. 1992; 360: 588-591. Ref.: https://goo.gl/xSyP6r
- Sacco A, Mourkioti F, Tran R, Choi J, Llewellyn M, et al. Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell.2010; 143: 1059-1071. Ref.: https://goo.gl/Li97aB
- Chandrasekharan K, Yoon JH, Xu Y, deVries S, Camboni M, et al. A human-specific deletion in mouse Cmah increases disease severity in the mdx model of Duchenne muscular dystrophy. Sci Transl Med. 2010; 2: Ref.: https://goo.gl/ksFNWv
- Bodor M, McDonald CM. Why short stature is beneficial in Duchenne muscular dystrophy. Muscle Nerve. 2013; 48: 336-342. Ref.: https://goo.gl/vQFJpG
- Hoen PA, de Meijer EJ, Boer JM, Vossen RH, Turk R, et al. Generation and Characterization of Transgenic Mice with the Full-length Human DMD Gene. J Biol Chem. 2008; 283: 5899-5907. Ref.: https://goo.gl/yELG4v
- INNES JR. Myopathies in animals; a record of some cases including progressive muscular dystrophy (pseudo-hypertrophic) (dog), “weisses Fleisch” (lamb), neuropathic muscular atrophy (sheep) and lymphocytic/histiocytic myositis, neuritis, radiculitis (dog). Br Vet J. 1951; 107: 131-143. Ref.: https://goo.gl/UXKGKh
- Sharp NJ, Kornegay JN, Van Camp SD, Herbstreith MH, Secore SL, et al. An error in dystrophin mRNA processing in golden retriever muscular dystrophy, an animal homologue of Duchenne muscular dystrophy. Genomics. 1992; 13: 115-121. Ref.: https://goo.gl/QCSR8f
- Cooper BJ, Valentine BA, Wilson S, Patterson DF, Concannon PW. Canine muscular dystrophy: confirmation of X-linked inheritance. J Hered. 1988; 79: 405-408. Ref.: https://goo.gl/rPTEK1
- Walmsley GL, Arechavala-Gomeza V, Fernandez-Fuente M, Burke MM, Nagel N, et al. A Duchenne muscular dystrophy gene hot spot mutation in dystrophin-deficient Cavalier King Charles spaniels is amenable to exon 51 skipping. PLoS One. 2010; 5: e8647. Ref.: https://goo.gl/iwbE3Y
- Nakamura K, Fuji W, Tsuboi M, Tanihata J, Teramoto N, et al. Generation of muscular dystrophy rat with CRISPR/Cas system. Sci Rep. 2014; 4: Ref.: https://goo.gl/KZNHY8
- Larcher T, Lafoux A, Tesson L, Remy S, Thepenier V, et al. Characterization of dystrophin deficient rats : a new model for Duchenne muscular dystrophy. Plos One. 2014; 9: e110371. Ref.: https://goo.gl/kMuk87
- Chen Y, Zheng Y, Kang Y, Yang W, Niu Y, et al. Functional disruption of the dystrophin gene in rhesus monkey using CRISPR/Cas9. Hum Mol Genet.2015; 24: 3764-3774. Ref.: https://goo.gl/y7uuSG
- Klymiuk N, Blutke A, Graf A, Krause S, Burkhardt K, et al. Dystrophin-deficient pigs provide new insights into hierarchy of physiology derangements of dystrophic muscle. Hum Mol Genet. 2013;22: 4368-4382. Ref.: https://goo.gl/LnqtKi